Secuenciación de PRRSV y su uso en la práctica
05-mar-2018 (hace 7 años 28 días)
El síndrome reproductivo y respiratorio porcino (PRRS) sigue siendo, con mucho, la enfermedad con mayor impacto económico en la industria porcina y su control dista de ser satisfactorio. Una mejor y más completa imagen de la variación del virus PRRS y la monitozación de la circulación de nuevas cepas dentro de una determinada área/país/continente ciertamente ayudaría a los veterinarios y productores a implementar programas de control y posiblemente de erradicación. Por este motivo, a partir de finales de 1990, la secuenciación del virus PRRS comenzó a estar disponible en todo el mundo, principalmente en América del Norte, Europa y el sudeste de Asia.
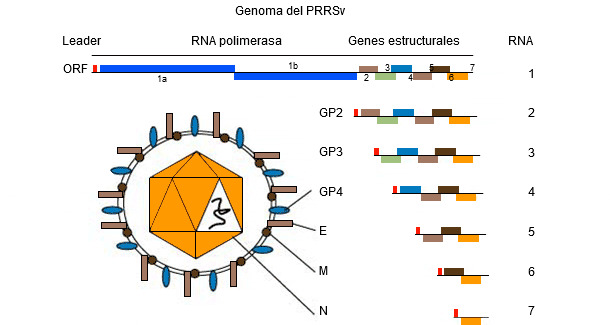
El genoma del virus PRRS (imagen 1) consiste en una molécula de ARN monocatenario que lo hace propenso a "cometer errores" (mutaciones genéticas) durante su replicación en el huésped. Esta "tendencia a cometer errores" resulta en la presencia en el campo de diferentes cepas de virus PRRS, todos únicos en su propia secuencia genética. Entre los veterinarios de campo y los investigadores sigue habiendo un debate sobre si estas diferencias en las secuencias contribuyen a "comportamientos diferentes" (ya sean clínicos-patológicos o inmunológicos).
Conceptos básicos de la secuenciación del PRRS
La secuenciación del virus se realiza a partir de productos de PCR procedentes de muestras de campo (sueros, tejidos, fluidos orales) obteniendo la lectura de nucleótidos generalmente de algunos fragmentos del genoma del ARN viral (ver figura 2) en determinadas regiones objetivo - ORF (Open Reading Frame) y luego se compara el porcentaje de homología mediante el análisis filogenético realizado utilizando softwares específicos. El resultado de este proceso proporciona el grado de similitud (homología) entre las diferentes cepas de virus PRRS. Usando softwares de visualización gráfica, también se puede obtener un dendograma (o "árbol filogenético") que muestra el grado de relación con la secuencia del virus de referencia (ver figura 3).

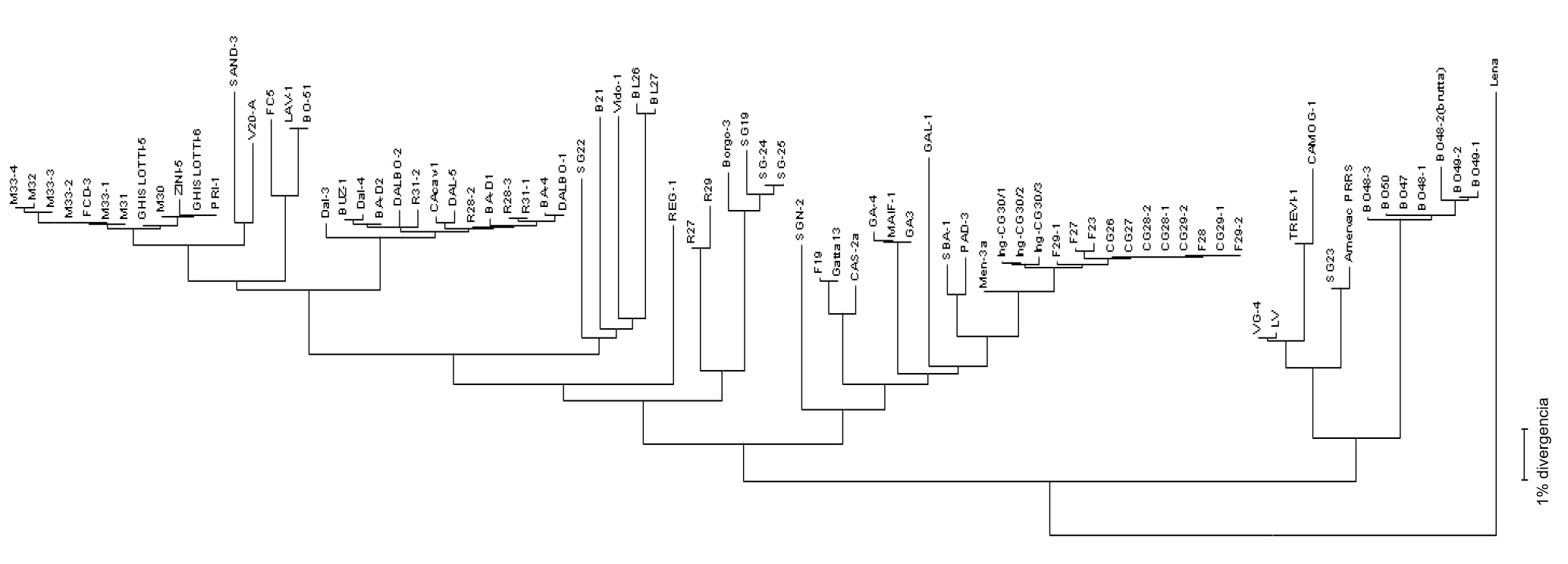
El genoma del PRRSV codifica al menos 10 ORF. Los más comúnmente utilizados para la secuenciación —aunque representan solo el 4% y el 3% respectivamente del genoma completo, son ORF5 (que codifica la proteína E no glicosilada) y ORF7 (que codifica la proteína de nucleocápside (N)). ORF5 representa una región más variable, mientras que ORF7 representa una región más conservada. Debido a esto, el mismo grado de variación (por ejemplo una variación del 5%) que se encuentre en ORF7 es más "dramática" en términos de cambio genético, en comparación con la misma variación en ORF5. La interpretación de las similitudes (es decir, si los virus están relacionados o no) requiere mucha más información adicional, ya que la tasa de cambio genético puede ser muy variable.
Es extremadamente importante mantener un archivo de registro de todas las secuencias identificadas unívocamente, anotando cuidadosamente la fecha, el tipo de granja (sitio 1-2-3), el flujo porcino, la ubicación (latitud / longitud del GPS) y el origen de la secuencia (tipo de animal/tejido/muestra). A día de hoy, nuestra base de datos de secuencias de virus PRRS abarca más de 1300 secuencias de ORF7 desde 2002. Para interpretar y dar sentido a las diferencias, es aún más importante relacionar secuencias individuales con eventos clínicos como el número de cerdas abortadas y la mortalidad predestete en Sitios 1 o la tasa de mortalidad en los sitios 2 y 3.
Preguntas prácticas
Las preguntas frecuentes de productores y veterinarios son:
- Las diferencias genéticas que se observan entre las secuencias ¿representan la variación normal de una sola cepa de PRRS en una granja/sistema, o representan múltiples cepas diferentes presentes al mismo tiempo, o en un corto espacio temporal, en una granja/sistema?
- ¿Lo que estoy teniendo ahora es un "nuevo brote" causado por una "nueva cepa" o es una recirculación?
Para responder a estas preguntas debemos acordar el grado aceptado de homología de dos cepas virales recogidas dentro de un cierto período de tiempo (¿12-24 meses?). En otras palabras, el punto de corte de similitud. Un 97-98% de homología en la secuencia (o una diferencia de 2-3%) es un valor generalmente aceptado. Según mi experiencia, es bastante difícil ver un cambio superior al 2% en una "población cerrada clínicamente estable" (una población convencional de cerdas o un flujo de cerdos) ya que lo que observamos es que obtenemos la "misma cepa" durante un período de hasta 3 años en una población única clínicamente estable. En el polo opuesto, cada vez que notamos una actividad consistente de PRRS, se recupera una cepa "nueva" y filogenéticamente diversa (90% de homología o menos). Lamentablemente, no sabemos con certeza si estas grandes diferencias que a veces observamos, son el resultado de un cambio repentino en el virus/mutación (improbable en mi opinión personal) o la introducción de una nueva cepa. Lo que está claro y bien aceptado es el hecho de que la similitud/diversidad genética no es de ninguna manera predictiva de similitud inmunológica (es decir, indicativa de inmunidad cruzada protectora) y no es en absoluto predictiva de la patogenicidad intrínseca (no dice si una cepa en particular es "buena" o "mala").
Las secuencias completas del genoma que están disponibles en la actualidad (lamentablemente aún más para fines de investigación que para el uso diagnóstico diario) sin duda ayudarán a responder esta pregunta.
Es muy importante analizar las nuevas secuencias de virus PRRS contra un amplio conjunto de referencia que represente la granja, el sistema y la región, así como las secuencias de las vacunas comerciales disponibles (esto permitirá diferenciar entre cepas de campo y vacunales). En este momento todavía estamos usando un software de código abierto administrado por la Universidad de Padova para construir nuestros árboles filogenéticos y mantenerlos organizados por flujo de cerdos dentro del total de nuestro sistema de producción; en un futuro cercano podríamos unirnos a otros dos programas informáticos "ad hoc" (Bioportal de la Universidad de Davis-California y CLASSIFARM-PATH de IZSLER (Brescia, Italia) que tendrán un conjunto mucho más grande de secuencias para comparar y permitir una mejor comprensión del virus PRRS circulante en Italia y posiblemente en la UE.
Agradecimientos: Gracias a la Prof. Michele Drigo (UNI-PD) por la interesante discusión y la revisión de este documento.