El desafío de la clasificación taxonómica intraespecífica del PCV2
27-may-2016 (hace 8 años 10 meses 6 días)
PCV2 ha emergido como una de las infecciones víricas más devastadoras en producción porcina, causando un impacto económico relevante por las pérdidas directas y los gastos en las estrategias de control. PCV2 es el agente etiológico del síndrome de adelgazamiento post-destete (PMWS), hoy en día conocido como enfermedad sistémica por PCV2 (ES-PCV2, figura 1). Los estudios epidemiológicos y experimentales han evidenciado que la diversidad genética, caracterizada por oleadas globales de genotipos emergentes y la circulación de cepas recombinantes, puede afectar potencialmente la virulencia de PVC2. Quizá esta es la razón por la que la clasificación intraespecífica de PCV2 ha sido históricamente controvertida. En 2008, el consorcio europeo de enfermedades asociadas al circovirus porcino propuso una nomenclatura estandarizada para las definiciones genotípicas de PCV2 basadas en comparación de secuencias pareadas (www.pcvd.eu). El análisis de secuencias de nucleótidos del genoma de PCV2 completo y de la cápside (ORF2) definieron dos umbrales de distancia de 0,020 y 0,035, respectivamente. Cuando la distancia (p) entre dos secuencias era mayor que dichos valores, se asumía que las cepas pertenecían a dos genotipos distintos. Los resultados de dichos análisis reconocieron cuatro genotipos llamados PCV2a, PCV2b, PCV2c y PCV2d (también conocido como PCV2b mutante).

Figura 1. Cerdo de tres meses de vida con ES-PCV2. Nótese la columna vertebral marcada, indicativa de retraso en el crecimiento y la palidez (anemia).
Desde 2008, un gran número de secuencias de PCV2 han sido depositadas en el GenBank (www.ncbi.nlm.nih.gov) y se han propuesto varios nuevos genotipos, aunque no siempre han sido validados. Una proporción significativa de dichas secuencias tiene un origen recombinante y en algunos casos han estado recirculando con una prevalencia creciente en varios países asiáticos y Estados Unidos. En un artículo reciente (Franzo et al, 2015), un equipo internacional de investigadores volvió a analizar la taxonomía intra-específica de PCV2 y la definición genotípica para comprobar su validez actual, unificar la nomenclatura y evitar posteriores errores de interpretación.
Los resultados obtenidos indican que, con las secuencias actualmente disponibles de cápside y genoma de PCV2, se han violado varios de los supuestos de la metodología que se estableció hace casi una década para establecer los umbrales. Concretamente, no se ha cumplido el supuesto de la misma tasa de sustitución de nucleótidos entre los distintos genotipos de PCV2. Además, la definición de un único valor de corte para definir los genotipos de PCV2 parece complicada. Todavía más importante, los umbrales propuestos para la definición de genotipo carecerían indudablemente de sentido (figura 2). Estas evidencias, junto con la enorme cantidad de nuevas secuencias disponibles, tienen implicaciones importantes en la clasificación intraespecífica de PCV2. Por lo tanto, los umbrales aplicados desde 2008 para definir los genotipos de PCV2 no son aplicables a todas las cepas de PCV2 existentes en la actualidad, por lo que el método debe reconsiderarse.
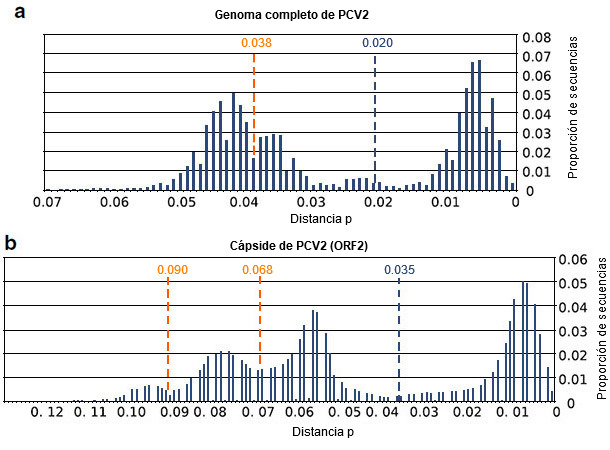
Figura 2. Resultados de la comparación de secuencias del genoma completo de PCV2 (a) y de secuencias de ORF2 (b). Se representa la proporción de secuencias comprendidas en un intervalo de distancia p de 0,01. También se indican los umbrales previamente propuestos para el genoma completo (0,020), la cápside (0,035), así como los umbrales resultantes del nuevo análisis (0,038, 0,068 y 0,090).
Como consecuencia se propone un método alternativo para determinar el genotipo de las cepas de PCV2 de forma inequívoca. El método sugerido tiene en cuenta la existencia de varias cepas recombinantes que, de hecho, pertenecen a más de un genotipo. Considerando la alta tasa de recombinación descrita, se ha preferido una clasificación basada en el gen ORF2. Con el objetivo de ofrecer un método de clasificación inequívoca, se seleccionaron varias secuencias de referencia cuya clasificación era clara según los métodos filogenéticos. Esto permitió definir cuatro genotipos utilizando tanto la reconstrucción filogenética como cuarenta y siete posiciones de nucleótidos marcadores específicos para cada genotipo. Estas posiciones de marcadores caracterizaban cada genotipo de un modo consistente (>95%) y podían ser utilizadas como referencia para asignar una secuencia problema a determinado genotipo. El método de clasificación propuesto, además de ser robusto, es muy rápido y fácil de llevar a cabo.
La clasificación taxonómica de PCV2 sigue siendo un desafío. Los resultados obtenidos confirman y validan la variabilidad de las secuencias virales y la elevada frecuencia de recombinación intra- e inter-genotipo entre las cepas de PCV2 resaltando la dificultad para establecer una definición de genotipo inequívoca para este virus. Además, la posibilidad de la aparición de nuevos genotipos de PCV2 en el futuro es muy elevada, y puede hacerse mucho más necesario tener nuevos, y robustos, métodos de genotipado.
La clasificación de PCV2 en genotipos es relevante desde un punto de vista práctico. Además, todas las vacunas disponibles en el mercado europeo y norteamericano están basadas en el genotipo PCV2a, aunque los que tienen más prevalencia son los PCV2b y PCV2d. Pese a que se ha demostrado un nivel significativo de protección cruzada entre estos tres genotipos, sería interesante evaluar si la eficiencia vacunal es equivalente frente a todos estos genotipos. Por otra parte, hasta el momento algún estudio filogenético publicado indica que pueden producirse ciertos cambios en aminoácidos en secuencias de PCV2 procedentes de explotaciones vacunadas y no vacunadas. De hecho, se ha especulado la posibilidad de que en el futuro se puedan generar virus mutantes debido a la presión de vacunación. Por lo tanto es clave una monitorización genotípica adecuada para evaluar la eficiencia vacunal.