Seguridad y residuos
19-jun-2012 (hace 12 años 10 meses 8 días)
En la Parte III de un expediente de registro se describen todos los aspectos del medicamento relacionados con la inocuidad del mismo. El objetivo fundamental de esta parte consiste en mostrar los riesgos potenciales derivados de la utilización del medicamento en las condiciones de uso propuestas.
Se deben tener en cuenta no sólo los efectos tóxicos del medicamento sobre el animal, sino también sobre la persona que administre el medicamento y sobre el medio ambiente. En el caso de medicamentos para a animales destinados a consumo humano, también deben incluirse en este apartado los correspondientes estudios de establecimiento del tiempo de espera.
Toda esta información se estructura en los siguientes apartados:
Farmacología y farmacocinética
En este apartado se describen los mecanismos mediante los cuales se producen los efectos terapéuticos pero también los efectos tóxicos. Estos efectos nocivos pueden producirse a partir de una determinada dosis o a partir de unos determinados niveles plasmáticos. Para aportar información farmacológica y farmacocinética es frecuente acudir a publicaciones científicas en las que se aportan datos de las sustancias activas, ya sea en estudios realizados en animales de laboratorio o en las especies de destino del medicamento. También se suelen incluir estudios farmacocinéticos llevados a cabo con el medicamento que se pretende registrar.
Esta información se refleja en los epígrafes del mismo nombre de la Ficha Técnica del producto (FT).
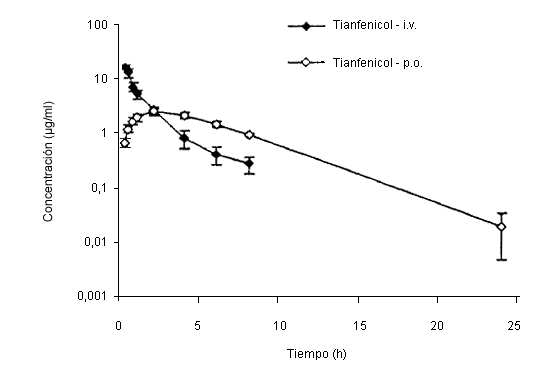
Concentración plasmática de tianfenicol tras administración oral e intravenosa.
Datos toxicológicos
Se aportan estudios o datos de toxicidad aguda y crónica, mutagenicidad y carcinogenicidad, inmunotoxicidad, propiedades microbiológicas de los residuos (en caso de tratarse de un antibiótico) y observaciones sobre el uso terapéutico en seres humanos. Normalmente se trata de datos bibliográficos.
También se incluyen estudios de tolerancia del medicamento en las especies de destino, que se suelen realizar administrando dosis normales y superiores a las normales para detectar posibles intolerancias del medicamento.
Los resultados de los estudios de tolerancia se reflejan en el apartado “Sobredosificación” de la FT.
Seguridad para el usuario
En esta sección se comentarán los efectos encontrados en las secciones anteriores, que se relacionarán con el tipo y grado de exposición humana al medicamento con objeto de formular las correspondientes advertencias para el usuario y demás medidas de gestión del riesgo, descritas en el apartado.
“Precauciones específicas que debe tomar la persona que administre el medicamento a los animales” de la FT.
Ecotoxicidad
Se aportan estudios que permiten la evaluación los efectos nocivos para el medio ambiente que pueda producir el medicamento. En concreto, se estudia el grado en que el principio activo o sus metabolitos entran en contacto con el medio ambiente, en función de la dosis propuesta, la especie de destino, pauta de utilización del medicamento (medicación individual o colectiva), la posibilidad de paso al medio ambiente mediante excreción, persistencia de las excretas y eliminación de los desechos o del producto no utilizado.
Estos estudios pueden dar lugar a recomendaciones especiales en el apartado “Precauciones especiales para la eliminación del medicamento veterinario no utilizado o, en su caso, los residuos derivados de su uso” de la FT.
Estudios de residuos
La realización de estudios de residuos, o de establecimiento del tiempo de espera, es una parte fundamental de un expediente de registro de un medicamento destinado a animales de abasto, ya que se trata de estudios costosos, que implican el sacrificio de un número considerable de animales y su resultado puede condicionar el éxito comercial del medicamento.
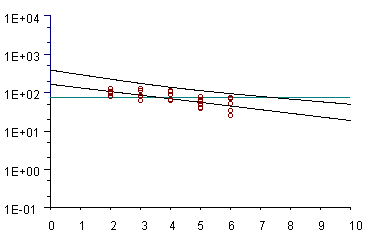
Gráfica de regresión de los valores de residuos (µg/ml) obtenidos en carne para un producto respecto a su LMR (línea verde) en el tiempo (días). La línea superior marca el margen de seguridad (95% de confianza).
Como norma general, los estudios de residuos se realizan con 4 grupos de 4 animales cada uno. Se debe realizar un estudio por especie de destino. Los animales son tratados según la dosis y pauta propuestas y se sacrifican lotes de 4 animales a diferentes tiempos. Se toman muestras de los diferentes tejidos diana (hígado, riñón, músculo, piel, grasa y en caso de inyectable, punto de inyección) y se determina en las mismas los niveles de la sustancia activa. A partir de la recta concentración/tiempo y mediante un tratamiento estadístico adecuado, se determina el tiempo de supresión, que permitirá asegurar que los niveles de residuos se hallen por debajo del Límite Máximo de Residuos (LMR) en todos los animales tratados.

Puntos de inyección con necrosis y restos de antibiótico.
Existen guías comunitarias que dan pautas para la realización de estos estudios.
La legislación vigente no permite comercializar medicamentos destinados a animales para consumo humano, cuyas sustancias activas no tengan establecido un LMR, que está publicado en el Reglamento 37/2010 de la Comisión de 22 de diciembre de 2009.
Es importante destacar que el tiempo de espera, al igual que la farmacocinética, están en gran medida influenciados por la forma farmacéutica y la composición cuali-cuantitativa de cada medicamento, es decir, de los excipientes que acompañen a la sustancia activa así como de las características físico-químicas de ésta (formas de cristalización, diferentes sales o tamaño de partícula).
Estos estudios darán lugar a los “Tiempos de espera” de la FT.